文章展示

實驗技術:WES+16S rDNA 測序 + 甲基化芯片
期刊:PNAS
時間:2019.12.26
研究路線
主要結果
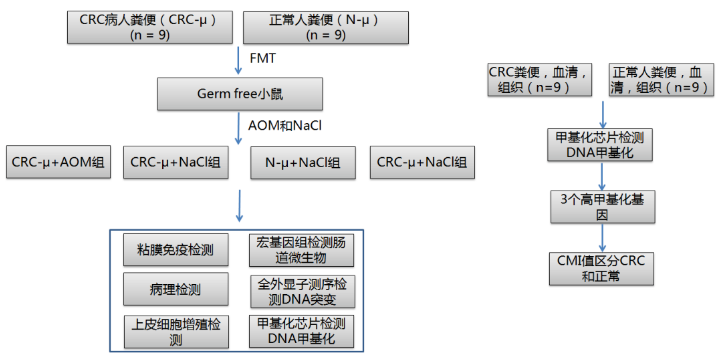
FMT 移植小鼠:結腸組織中與生物異常相關的組織學改變
FMT 后,N- μ 組表現正常。而 CRC- μ 或 AOM 單獨處理,以及 CRC-μ+AOM 處理組都表現出了輕微的炎癥反應,以及異常隱窩病灶。通過 mRNA 細胞因子定量評估,AOM 治療傾向于放大這種刺激作用。此外,對整個組織學檢查腸粘膜髓細胞的半定量評估顯示,與 N - μ 相比,CRC- μ 的粘膜髓細胞數量有增加的趨勢,但在任何時間點都沒有達到統計學意義。
圖 A -G: 組織病理學變化;圖 H: 對細胞增殖,病理癌變的的影響;圖 I:ACF 的數量;圖 J:粘膜炎癥變化;圖 K: 腸粘膜髓細胞的變化。
小鼠體內 FMT 后的微生物區系特征變化
為了評估微生物群落對觀察到的粘膜表型差異的影響,對小鼠糞便樣本進行 16S rDNA 測序分析,結果顯示,在基線水平下,與 N - μ 組相比,CRC-μ(n = 9 個供體)組的糞便微生物群中梭桿菌、帕維單胞菌、丁酸弧菌、和低比例的 Ruminococcus,雙歧桿菌、真細菌和 Lachnospira。隨訪期間,各組的 Coccoides, Clostridium leptum, 和 Bifidobacterium 均出現中度下降。此外還發現,組織學改變與起到抗炎作用的細菌含量減少相關(Faecalibaterium,Eubacterium 以及厚壁菌門的種屬)。
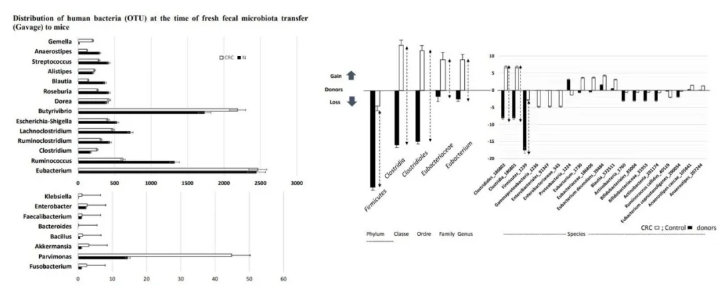
FMT 對小鼠 DNA 突變的影響
為了分析 FMT 對宿主進行 DNA 修飾的影響,研究人員對小鼠結腸黏膜組織進行全基因組測序,覆蓋 24000 個基因中的 220,000 個外顯子。小腸組織中整體外顯子 / 內含子水平的突變發生率明顯高于脾臟組織。在結腸黏膜中,N- μ 組在外顯子或內含子的 DNA 發生改變要比 CRC- μ 組低,但并沒有達到顯著性。此外,給予 CRC-μ+AOM 組的小鼠顯示出高水平的外顯子或內含子的 DNA 改變。為了分析 CRC- μ 誘發的局部致癌潛力,作者繼續對以下選擇的基因通路進行了深入分析:Wnt 和 catenin(19 個)、Notch(4 個)、PPAR(3 個)、SMAD(2 個)、TGF- inhibitor 2 個基因,ACRV 2 個基因,DKK 4 個基因,TCF 2 個基因,MYC 1 個基因,SOCS 1 個基因。顯著的變化發生在關鍵的 Wnt 通路基因中(11 個 Wnt 基因中存在 indel 和單核苷酸多態性),CRC- μ 組和 N - μ 組之間沒有顯著差異。根據結腸粘膜或脾臟樣本中單核苷酸的總 DNA 變化,對動物進行亞群分析,表明基因突變與微生物類型(N 或 CRC) 或給小鼠的治療(生理鹽水或 AOM) 有關。
圖 A:CRC 糞便處理組比對照組,在 exon 和 intron 上的 SNP 位點要多,但是不顯著;圖 B:基于 SNP 的 PCA;圖 D:不同組間 Wnt 基因突變率比較,AOM +CRC- μ 組高
FMT 對小鼠 DNA 甲基化的影響
在小鼠結腸粘膜組織,CRC- μ 相對于 N - μ 可以誘發更大的表觀遺傳改變。N-μ + NaCl 甲基化高于 CRC-μ + NaCl 高于 CRC-μ + AOM 組;CRC-μ + AOM 組的為甲基化探針數比 N -μ + NaCl 組高 11%,CRC-μ 和 AOM 處理后,完全甲基化的探針較高。
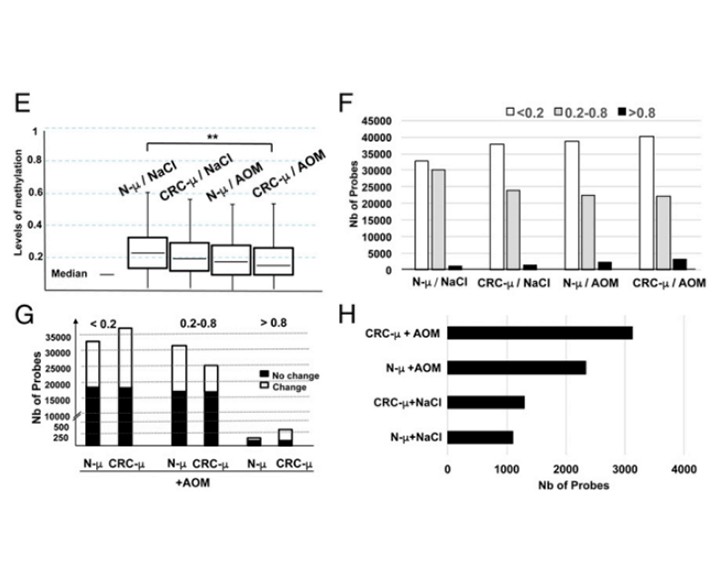
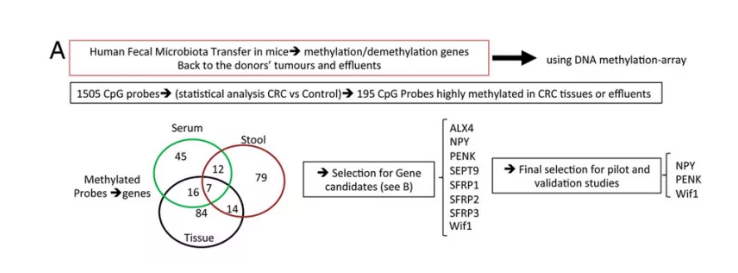
結腸癌病人樣本中的基因甲基化
通過檢測血液,糞便,CRC 組織和排泄物(n = 9) 和正常組排泄物(n = 9) 的甲基化,從高甲基化的基因上,選出 8 個在所有樣本中都一致的基因(Wif1-regulating gene and SEPT9, SFRP1,2,3,PENK, NPY, and ALX4 genes),確定了 3 個基因進行驗證。三個基因和一個看家基因的 CMI 值可以區分 CRC 和正常人,specificity and the sensitivity of CMI > 2 in blood was 95 and 59%。多因素分析顯示 CMI 是 CRC 診斷的獨立危險因素,包括糞便免疫化學血液檢測。
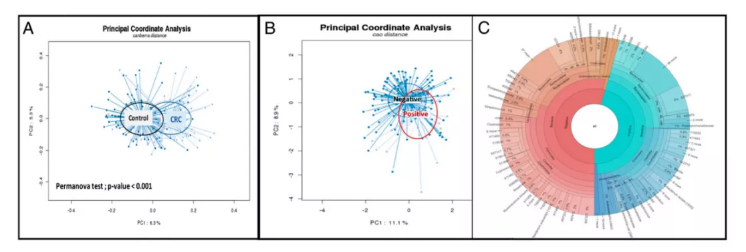
微生物組成與 CMI 的關系
根據基因的 CMI 值可以把群體分成兩類,CMI 大于 2 和 CMI≤2。宏基因組分析顯示,在 CMI > 2 患者(n = 53) 和 CMI≤2(n = 90) 患者中,包括幾種 Parvimonas(單胞菌)在內的 20 種細菌在豐度上存在差異。
結論
CRC 相關的微生態失調導致宿主基因甲基化,相應的 CMIs 和相關細菌可能是 CRC 的 biomarkers。
參考文獻:Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. Sobhani et al. Proc Natl Acad Sci U S A (2019)
更多伯豪生物服務

