
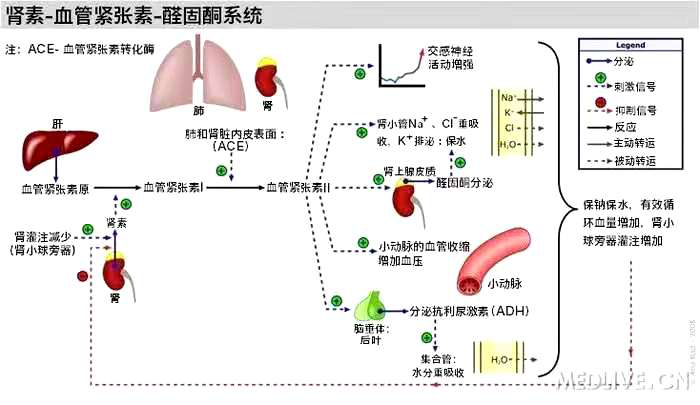
根據(jù) 2020 年 9 月出版的《中國心血管健康與疾病報告 2019 概要》,我國心血管病患病率處于持續(xù)上升階段,推算心血管病現(xiàn)患人數(shù)為 3.30 億,其中高血壓 2.45 億,也就是說,每 5 個成人中就有 1 個高血壓患者。高血壓又是心血管病死亡里的頭號致死因素,每 4 例死亡中就有 1 例死于高血壓。

高血壓定義
根據(jù) 2020 年 5 月 6 日國際高血壓學(xué)會(ISH)發(fā)布的《ISH2020 國際高血壓實踐指南》,在連續(xù)多次重復(fù)測量后,診室血壓≥140/90mmHg 即可診斷為高血壓。

單基因遺傳性高血壓
是指由單一基因突變導(dǎo)致的高血壓,為常染色體顯性或隱性遺傳,多早年發(fā)病,預(yù)后較差。對遺傳性疾病進行基因檢測和診斷,有助于患者及親屬的早期診斷和鑒別,還對治療策略的制定、遺傳篩查以及選擇性生育等有重要的作用。

醛固酮
醛固酮是人體內(nèi)重要的鹽皮質(zhì)激素,促進腎臟對鈉的吸收,維持水平衡。但醛固酮過多是引起高血壓的一個常見但未被認識的原因,其引起的頑固性高血壓,患者心臟、腎臟等高血壓靶器官損害更為嚴重。
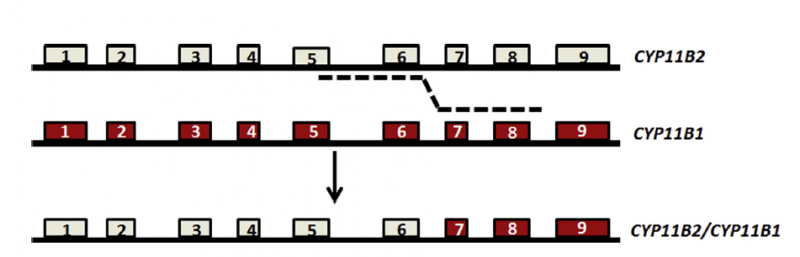
家族性醛固酮增多癥的分類
“家族性醛固酮增多癥”是由于遺傳因素而產(chǎn)生的疾病。根據(jù) 2020 年 9 月 30 日發(fā)布的《原發(fā)性醛固酮增多癥診斷治療的專家共識(2020 版)》、2019 年 3 月發(fā)布的《單基因遺傳性心血管疾病基因診斷指南》,因致病基因不同,家族性醛固酮增多癥分為 4 個亞型:FHA I、FHA II、FHA III 和 FHA Ⅳ。例如 FHA I 型是由于 CYP11B1 基因和 CYP11B2 基因重組所產(chǎn)生的嵌合基因引起醛固酮合成調(diào)控的改變,導(dǎo)致糖皮質(zhì)激素可抑制性醛固酮增多癥。通過進行基因檢測,可確定分型,針對不同分型采取正確的治療方案。
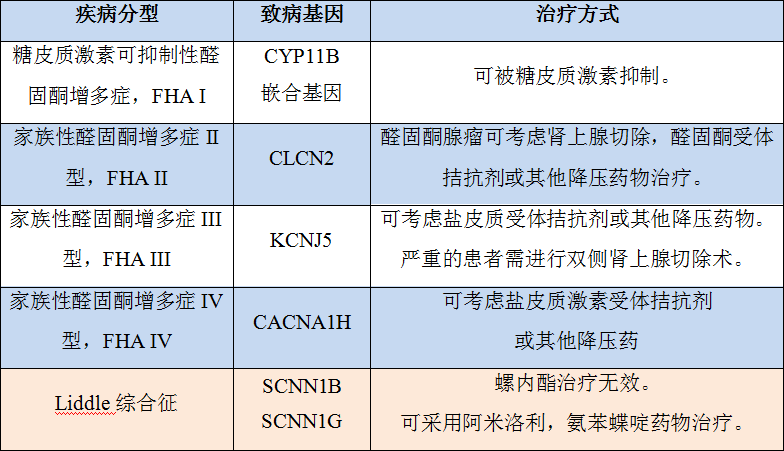
家族性醛固酮增多癥的 篩查與治療
前面提到,按照產(chǎn)生機制不同,家族性醛固酮增多癥分為四種類型。通過基因檢測,進行基因分型,即可判斷出疾病類型。四種類型的遺傳方式都是常染色體顯性遺傳。
產(chǎn)生機制不同,治療方式不同,如 FHA I 的顯著特點是可被糖皮質(zhì)激素抑制,其他幾種,也因致病基因不同,需采取不同的治療方式。
假性醛固酮增多癥:Liddle 綜合征
Liddle 綜合征,其臨床表現(xiàn)酷似原發(fā)性醛固酮增多癥,但是致病機制不同,這是由編碼上皮細胞鈉通道蛋白 β、γ 亞單位的 SCNN1B 和 SCNN1G 基因突變導(dǎo)致。通過基因檢測可以區(qū)分 Liddle 綜合征和“醛固酮增多癥”,從而采用正確的治療方式,得到好的治療效果。基因診斷是 Liddle 綜合征的金標準。
治療藥物與基因
提到高血壓,很多人的反應(yīng)就是要吃降壓藥。但同是降壓藥,有些人服用有效,而有些人卻沒有降壓效果,這就和我們?nèi)梭w的基因有關(guān)系了。治療藥物進入體內(nèi)后,藥物在體內(nèi)代謝、轉(zhuǎn)運和藥物作用靶點基因的遺傳變異及其表達水平的變化,可影響藥物在體內(nèi)的濃度和敏感性,導(dǎo)致藥物反應(yīng)個體產(chǎn)生差異。例如,華法林是一種常見的口服抗凝劑,臨床用于血栓的預(yù)防和治療。人 CYP2C9 基因上特定突變會影響藥物代謝酶的活性,從而影響華法林在體內(nèi)的代謝。
因此,為了得到較好的治療效果,在進行疾病基因分型的前提下,還需要進行與治療藥物相關(guān)的基因多態(tài)性檢測。
疾病研究的深入、基因探索的進步,使得以前較少發(fā)現(xiàn)的疾病浮出了水面,家族性醛固酮增多癥引起的高血壓病例數(shù)也在逐步增多,罕見病也不再罕見,伯豪生物和重慶伯豪臨檢實驗室團隊,已經(jīng)開發(fā)了家族性醛固酮增多癥基因檢測產(chǎn)品,通過外周血的取樣檢測,即可進行相關(guān)基因的分型,完成疾病的分類、治療藥物基因檢測。 詳情請聯(lián)系:
上海伯豪生物技術(shù)有限公司:
地址:上海張江高科技園區(qū)李冰路 151 號
電話:8621-51320288
信箱:chunxiu_zhang@shbio.com
重慶伯豪醫(yī)學(xué)檢驗實驗室:
地址:重慶市大渡口區(qū)翠柏路 101 號 5 幢 16 樓
電話:8623-68870868
信箱:xinghua_xu@shbio.com
參考文獻:
1. 中國心血管健康與疾病報告 2019 概要,中國循環(huán)雜志,2020,35(9):833-854。
2. 原發(fā)性醛固酮增多癥診斷治療的專家共識(2020 版)。中華內(nèi)分泌代謝雜志,2020,36(9):727-736。
3. 單基因遺傳性心血管疾病基因診斷指南。中華心血管病雜志,2019,47(3):175-196。
4. 彭慧,胡曉軍,陳蘇等。醛固酮合酶基因多態(tài)性與老年原發(fā)性高血壓的關(guān)系。中國老年學(xué)雜志,2010,10:1326-1327。
5. 張霞,王衛(wèi)慶。醛固酮與頑固性高血壓。上海交通大學(xué)學(xué)報(醫(yī)學(xué)版),2010,30(5):511-514。
6. 宋宮儒。醛固酮過多產(chǎn)生是引起高血壓的一個常見但未被認識的原因。血管與腔內(nèi)血管外科雜志,2020.05.27。
7. Xu L,Xia W, Wu X, et al. Chimeric CYP11B2/CYP11B1 causing 11 β-hydroxylase deficiency in Chinese patients with congenital adrenal hyperplasia. Steroids, 2015, 101: 51-55.
8. Scholl UI, Nelson- Williams C, Yue P, et al. Hypertention with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci USA, 2012, 109(7): 2533-2538.
9. Murthy M, Xu S, Massimo G, et al. Role for Germline Mutations and a Rare Coding Single Nucleotide Polymorphism Within the KCNJ5 Potassium Channel in a Large Cohort of Sporadic Cases of Primary Aldosteronism. Hypertension, 2014, 4: 783-789.
10. Scholl UI, Stolting G, Schewe J, et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet, 2018, 50(3): 349-452.
11. Scholl UI, Stolting G, Nelson- Williams C, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early onset hypertension with primary aldosteronism. Elife. 2015, 4: e06315.
更多伯豪生物人工服務(wù):

