空間轉錄組技術的出現,使研究人員了解不同轉錄本的空間位置信息。但是,目前的技術還達不到單細胞的分辨率水平。也就是說,探針在特定位置捕獲到的轉錄組是來源于多個細胞的,而且這些細胞可能來自一組異質性的細胞,而并非同一類型的。因此,在組織任何位檢測到的基因表達譜都可以被認為是來自多個不同細胞(相同類型細胞或不同類型細胞)來源的轉錄本的混合物。這也就意味著,即使轉錄產物的空間位置信息可以完全繪制成圖表,但是產生這種轉錄的細胞的類型和空間分布仍然很大程度上是未知的。
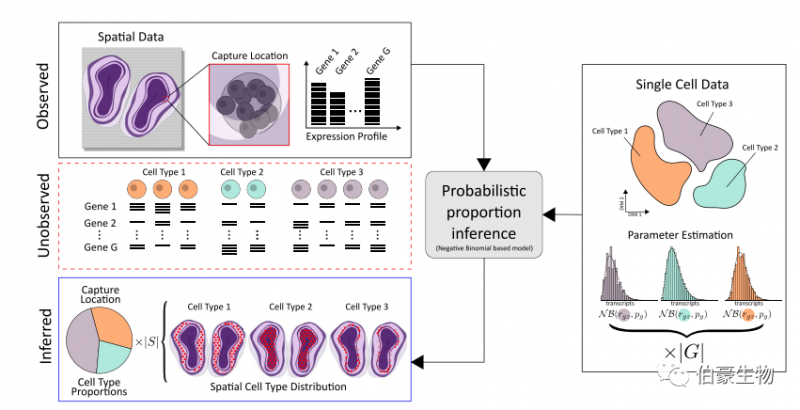
如上所述,空間轉錄組學技術面臨著一個困境,即知道轉錄本的位置,但不知道是哪個細胞產生了它們。相反,單細胞 RNA 測序可以將每個轉錄本與單個細胞聯系起來,但是關于這些轉錄本在組織中的位置的信息丟失了。鑒于這種互補的優勢和劣勢,結合兩種技術的數據來描繪細胞類型群體的空間信息是很好的選擇。
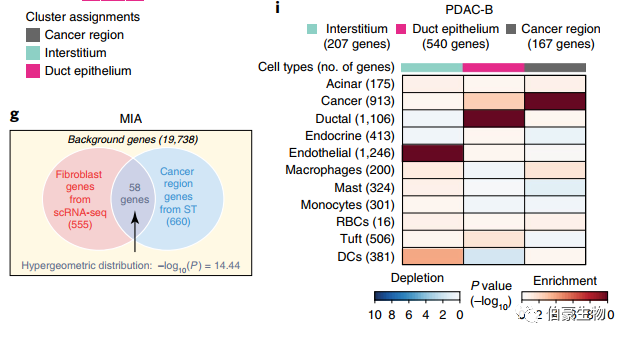
先前研究已經報道了,基于 bulk RNA-seq 數據來鑒定細胞類型的方法。發表在 nature biotechnology 上的一篇關于胰腺導管癌的單細胞和空間轉錄組的研究,基于 MIA(Multimodal intersection analysis)的算法也可以將細胞類型與空間位置對應起來。這種分析首先通過描述細胞類型特異性基因和組織區域特異性基因,然后確定它們的重疊是比偶然預期的高(富集)還是低(缺失)。在 scRNAseq 數據中,定義基因集的方法是,在每個細胞類型中,在標注到該細胞類型的細胞中,與其他細胞的表達量相比,其表達量在統計學上更高(P<10e?5)。對于 ST 數據,確定了每個空間區域相對于其他區域表達顯著更高的基因(P<0.01,雙尾 student t 檢驗)。通過 scRNA-seq 和 ST 提取的基因集,MIA 接下來計算每對細胞類型特異性和區域特異性基因集之間的重疊,并執行超幾何測試來評估顯著富集或缺失。[1]
發表在 COMMUNICATIONS BIOLOGY 雜志上的一篇文章提出了一種新的替代的基于模型的方法來整合單細胞 RNA 測序和空間轉錄組測序數據,它利用完整的表達譜而不是精選的一組標記基因。使用這種方法,研究人員對來自小鼠大腦和發育心臟的單細胞數據中的細胞類型空間映射到相應的組織切片上。并證實該方法優于其他方法 [2]。
在未來,單細胞測序聯合空間轉錄組測序會是全面解析樣本中細胞類型和空間位置信息的新方法,也是突破已知,探索未知的新技術領域。
參考文獻
【1】Moncada, R., Barkley, D., Wagner, F. et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol (2020) doi:10.1038/s41587-019-0392-8
【2】Alma Andersson1, Joseph Bergenstr?hle1 et al. Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. COMMUNICATIONS BIOLOGY (2020) doi.org/10.1038/s42003-020-01247-y
伯豪生物 - 空間轉錄組測序服務優勢
高質量切片: 切片、貼片經驗豐富,針對不同組織優化了解決方案。
流程化分析: 完善的分析流程,準確快速解析空間轉錄組數據。
標準化內控: 豐富的實操經驗構建了標準化的內控體系。
專業化團隊: 資深的技術團隊具有多年項目方案設計、實驗操作、售后分析等經驗。
全流程服務: 提供組織冷凍包埋、貼片、切片、透化、建庫測序及數據分析的全套服務。
更多伯豪生物人工服務:

