前面我們有介紹了利用 10x Space Ranger 軟件分析空間轉錄組原始數據得到可用于下游分析的矩陣和鏡像文件。今天來介紹一下怎么利用 Space Ranger 的結果文件進行后續分析,這里主要使用 Seurat 在進行下游分析。
先來回顧一下跑完 Space Ranger 得到哪些結果文件:
Outputs:
|
用 seurat 進行下游分析主要用到兩個結果文件。一個是 filtered_feature_bc_matrix.h5 文件,一個是 spatial 鏡像結果目錄。
安裝 R 包
由于 seurat 分析空間轉錄組的 R 包 satijalab-seurat 是在 GitHub 上的,如果我們需要直接安裝的話需要先安裝 R 包 devtools,然后利用 devtools 工具中的 install_github 來安裝 GitHub 上的 R 包。
安裝 devtools
install.packages('devtools')安裝 satijalab-seurat
devtools::install_github("satijalab/seurat",ref = "spatial")考慮到直接安裝 github 上的 R 包速度是很慢的,非常考驗網速,可能需要多次才能安裝成功,我們也可以直接下載安裝包,本地安裝。
# 下載https://codeload.github.com/satijalab/seurat/legacy.tar.gz/spatial# 安裝install.packages("satijalab-seurat-v3.1.5-351-g85610bc.tar.gz", repos = NULL, type = "source")
注意該 R 包還在開發中,不要和之前安裝的 seurat 包沖突。
數據準備
這里使用從 10x 官網下載的小鼠腦組織樣本 MouseBrain Serial Section 1 (Sagittal-Posterior)。
下載網址:https://support.10xgenomics.com/spatial-gene-expression/datasets
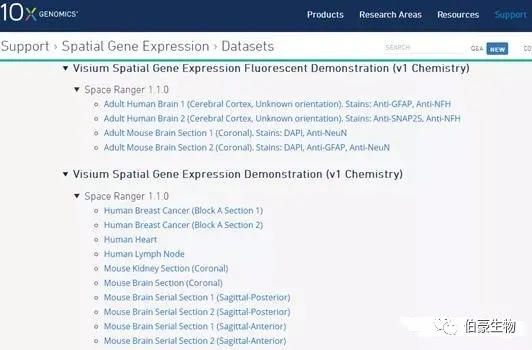
點擊選擇的樣本,下載兩個數據就行:
cell matrix HDF5 (filtered)和 Spatial imaging data
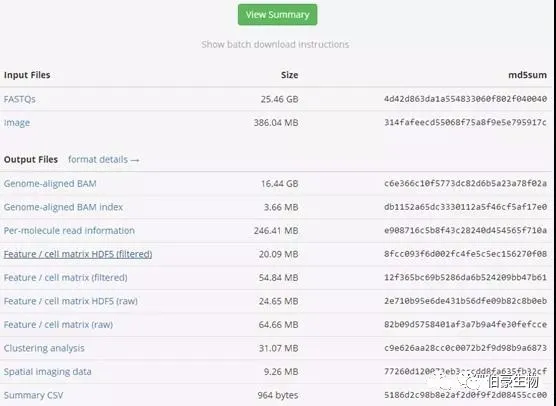
導入 R 包,讀取文件
library("Seurat")library("ggplot2")library("cowplot")library("dplyr")library("hdf5r")## 讀取矩陣文件name='Posterior1'expr <- "/pubj/ST_test/RNA/Sagittal-Posterior1/V1_Mouse_Brain_Sagittal_Posterior_filtered_feature_bc_matrix.h5"expr.mydata <- Seurat::Read10X_h5(filename = expr)mydata <- Seurat::CreateSeuratObject(counts = expr.mydata, project = 'Posterior1', assay = 'Spatial')mydata$slice <- 1mydata$region <- 'Posterior1' #命名# 讀取鏡像文件imgpath <- "/pubj/ST_test/RNA/Sagittal-Posterior1/spatial"img <- Seurat::Read10X_Image(image.dir = imgpath)Seurat::DefaultAssay(object = img) <- 'Spatial'img <- img[colnames(x = mydata)]mydata[['image']] <- imgmydata #查看數據An object of class Seurat32285 features across 3355 samples within 1 assayActive assay: Spatial (32285 features)
從 mydata 的輸出信息我們可以知道,這個樣本包含 3355 個 spot 點、32285 個基因。
基礎統計作圖
##UMI 統計畫圖plot1 <- VlnPlot(mydata, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata, features = "nCount_Spatial") + theme(legend.position = "right")plot_grid(plot1, plot2)
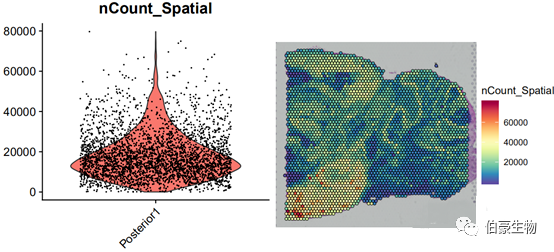
UMI 數大部分集中到 10000-20000 區間,不超過 80000,并且組織中高 UMI 數的區域主要集中在左下角。后面可以關注一下左下角區域的基因的表達和主要的細胞類型。
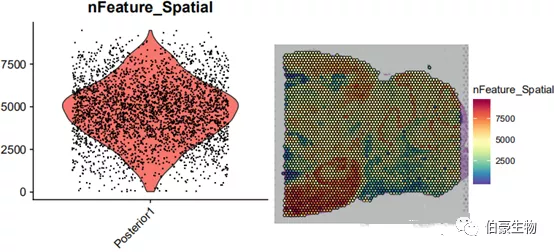
##gene 數目統計畫圖plot1 <- VlnPlot(mydata, features = "nFeature_Spatial", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata, features = "nFeature_Spatial") + theme(legend.position = "right")plot_grid(plot1, plot2)
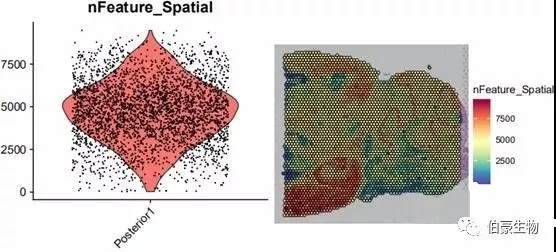
基因數目大部分處于 2500-7500 之間,結合 UMI 數據的分布可以發現 UMI 數目高的區域基因數也高,說明基因數和 UMI 數基本上是呈正相關的。
# 線粒體統計mydata[["percent.mt"]] <- PercentageFeatureSet(mydata, pattern = "^mt[-]")plot1 <- VlnPlot(mydata, features = "percent.mt", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata, features = "percent.mt") + theme(legend.position = "right")plot_grid(plot1, plot2)
注意如果是人的數據 pattern ="^mt[-] 改成 pattern ="^MT[-]
總體來說,這個樣本的線粒體比例不高,左邊中上區域有一處線粒體比例稍微高一點,后面也可以仔細研究一下這一塊區域到底是特定的細胞類型引起的還是組織活性的差異引起的。不過從這張圖我們還可以發現一個有意思的現象,基因和 UMI 高表達的區域往往線粒體比例更低。
數據過濾
做單細胞 RNAseq 我們都會根據 UMI、基因數、線粒體比例等進行過濾,那么做空間轉錄組數據分析其實我們也可以按這樣的方式來過濾。具體的過濾條件需要根據具體樣本數據來定,沒有固定的標準。
比如這個樣本我們可以設置過濾條件:
①基因數大于 200,小于 7500
②UMI 數大于 1000,小于 60000
③線粒體比例小于 25%
mydata2 <- subset(mydata, subset = nFeature_Spatial> 200 & nFeature_Spatial <7500 & nCount_Spatial> 1000 & nCount_Spatial <60000 & percent.mt < 25)mydata2An object of class Seurat32285 features across 2977 samples within 1 assayActive assay: Spatial (32285 features)
過濾后還剩 2977 個 spot 點。過濾后我們在繪制一下 UMI 分布圖。
plot1 <- VlnPlot(mydata2, features = "nCount_Spatial", pt.size = 0.1) + NoLegend()plot2 <- SpatialFeaturePlot(mydata2, features = "nCount_Spatial") + theme(legend.position = "right")plot_grid(plot1, plot2)

那么現在問題來了,過濾之后組織圖像里面缺了幾塊,顯得特別丑。那么我們到底應不應該過濾呢?過濾數據可以減少離群的點,減少對后面聚類結果的影響,不過濾數據可以讓組織圖像保持完整性,繪圖更好看一點,所以這個還真不好決斷。
數據歸一化
Seurat 官方推薦使用 sctransform 歸一化方法,它構建了基因表達的正則化負二項模型,以便在保留生物差異的同時考慮技術因素。
Sctransform 函數同時實現了 NormalizeData、ScaleData、FindVariableFeatures 三個函數的功能。
mydata <- SCTransform(mydata, assay = "Spatial", verbose = FALSE)基因表達可視化
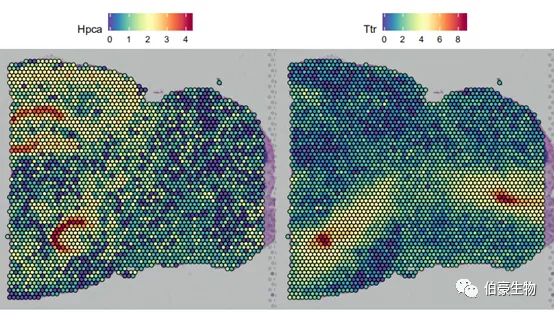
Seurat 的 SpatialFeaturePlot 功能擴展了 FeaturePlot,可以將表達數據覆蓋在組織組織上。這里展示的 Hpca 基因是一個強的海馬 marker,Ttr 是一個脈絡叢 marker。可以通過基因的表達分布來初步判斷一下海馬區和脈絡叢區處于組織切片的哪個位置。
SpatialFeaturePlot(mydata, features = c("Hpca", "Ttr"))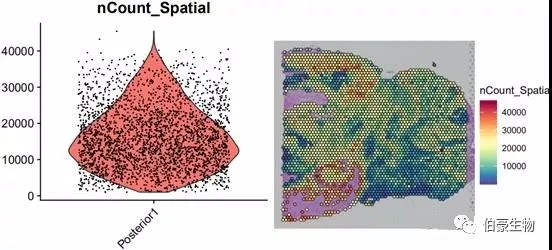
從結果的展示來看,這兩個 marker 基因的分布還是挺集中的,這也說明理由空間轉錄組數據來分析小鼠腦的不同區域的表達差異應該還是比較準確的。另外,海馬區的分布可以大概分成 3 大塊,從上之下首塊弧形區域似乎處于線粒體高表達區域,而下面一塊弧形區處于基因高表達區。后面可以把這三個不同區域的數據進行差異基因和功能的比較也許會發現一些有意思的東西。
降維、聚類和可視化
接下來利用 seurat 進行降維和聚類。先進行 PCA 降維,再選擇前 30 個維度進行聚類和 umap、tsne 降維。
mydata <- RunPCA(mydata, assay = "SCT", verbose = FALSE)mydata <- FindNeighbors(mydata, reduction = "pca", dims = 1:30)mydata <- FindClusters(mydata, verbose = FALSE)mydata <- RunUMAP(mydata, reduction = "pca", dims = 1:30)mydata <- RunTSNE(mydata, reduction = "pca",dims = 1:30)
tsne 展示結果:
Umap 展示結果:
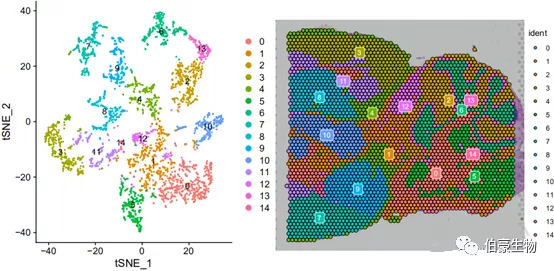
tsne 和 umap 兩種展示方式在這次分析里差別不是特別大,tsne 相對來說亞群與亞群之間分的更開,而 umap 則單個亞群位置更集中。這個時候我們也可以結合前面 marker 基因的表達分布圖來大概判斷一下每個亞群大概處于小鼠腦的那個區。
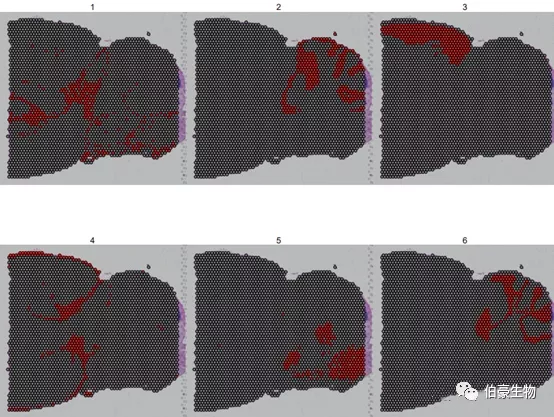
由于亞群的顏色比較接近,有時候不太好判斷,我們可以是 cells.highlight 來標記特定的亞群。
SpatialDimPlot(mydata, cells.highlight = CellsByIdentities(object = mydata, idents = c(1, 2, 3, 4,5, 6)), facet.highlight = TRUE, ncol = 3)
今天先分享到這,下期繼續!
更多伯豪生物人工服務:

