高通量測序技術的發展為人類疾病機制研究、遺傳病診斷、產前篩查與診斷、腫瘤診斷與治療等帶來了新的應用機遇。全外顯子組測序,是利用序列捕獲技術將全基因組外顯子區域的 DNA 捕捉并富集后,進行高通量測序的基因組分析方法。據估計,大約 85% 的人類遺傳變異集中在蛋白編碼區,因此對全部的蛋白編碼區(外顯子組)進行測序,可以快速、有效地鑒定人類疾病遺傳變異。外顯子區段大小約為全基因組的 1%,在相同的測序通量下,全外測序針對目標區段的測序深度以及覆蓋度都高于全基因組重測序,因此該技術對于發生在外顯子區的常見和罕見的染色體變異具有很高的檢測靈敏度。
全外測序與全基因組測序相比,不僅費用更低,數據的解讀也相對簡單。通過對疾病樣本進行全外測序,不僅能快速發現罕見遺傳疾病的致病基因,也能用于研究多基因引起的常見疾病,如癌癥、糖尿病、高血壓等,來揭示這些疾病的遺傳致病機理。全外測序是當前的熱點技術,極大地推動了疾病研究的進展。全外在疾病研究領域的廣泛應用和突出貢獻,2010 年入選《Science》雜志年度“十大科學進展”。小編將分別從①遺傳病,②復雜疾病,③腫瘤及個性化腫瘤疫苗方向介紹相關應用。
孟德爾遺傳病又稱單基因病,以往鑒定孟德爾遺傳病的致病基因大多利用連鎖分析結合候選定位克隆的方法,但耗時長,而且成功率低。2009 年報道了全外在孟德爾疾病致病基因定位的成果。發表于《Nature》的文章應用全外測序在 4 名弗里曼謝爾登綜合征患者的 DNA 中準確找出了致病基因(MYH3),表明了全外測序技術可以準確地找到罕見疾病的致病基因。隨后越來越多的孟德爾遺傳病的致病基因通過全外技術而被鑒定出來:米勒綜合征(DHODH)、Ohdo 綜合征(KAT6B)、陣發性運動障礙(PRRT2)、Bohring-Opitz 綜合征(ASXL1)、脊椎干骺端發育不良(KIF22)、先天性靜止性夜盲(LRIT3)等幾百種。
精選文獻
基于 Trios 家系的致病基因定位
樣本類型:全血
實驗設計:全外測序后根據遺傳規律分析遺傳類型:
常染色體隱性突變:篩選父母雜合,患者純合的突變。
復合雜合突變:是指同一基因的不同位點發生突變,這種突變是父母一方的某個基因的位點發生變異,但另一方在該基因的另外一個位點發生變異,然后該變異遺傳給子代,導致子代患病。
新生突變:一般指父母沒有攜帶致病基因,由于有害突變造成子女患病,可遺傳給下一代,是顯性突變。
常染色體顯性突變:篩選患者攜帶的突變,正常個體未攜帶的突變。
參考案例: 應用全外顯子組測序對 Holt-Oram 綜合征致病基因的定位【查看】
Holt-Oram 綜合征(HOS)即遺傳性心血管上肢畸形綜合征是一種常染色體顯性疾病,發病率為 1 /100000,其特征是上肢畸形和先天性心臟缺陷。位于染色體 12q24.1 上 TBX5 基因的改變已被認為 HOS 的主要原因,該基因突變可致基因轉錄功能降低而引起該綜合征。而某些病例無法通過 TBX5 突變來解釋。本研究為一例診斷為 HOS 的新生兒,有法洛四聯癥,拇指發育不全,面部發育不良和右耳道畸形。
基于大家系樣本的致病基因定位
樣本類型:全血
實驗設計:家系連鎖分析定位區段 + 全外測序篩選突變位點
樣本數:三代及以上家系樣本
參考案例: 應用全外顯子組測序和全基因組連鎖分析在非綜合征耳聾家系中鑒定 DMXL2 致病變異【查看】
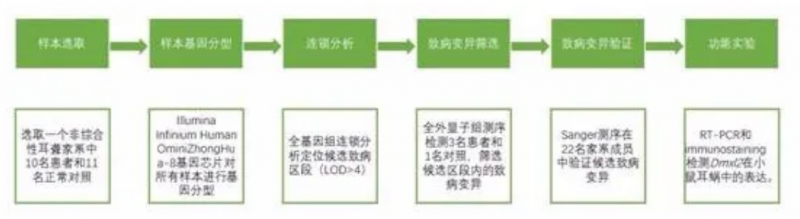
目前已知有超過 100 多個基因,其所含的致病變異會對聽覺系統造成不同的功能影響,并引起相應的聽力損失。但對非綜合征耳聾而言,依舊有超過 50 多相關位點的遺傳致病機制還未詳細闡明。本文利用全基因組 SNP 芯片及全外顯子組測序技術對一非綜合征耳聾家系中的 21 個樣本進行全基因組連鎖分析,并通過對個別家系樣本進行全外顯子組測序和對所有家系樣本進行 Sanger 測序來鑒定候選的致病變異。
參考文獻
[1] Clayton-Smith J, O'Sullivan J, Daly S, Bhaskar S, Day R,Anderson B,Voss AK, Thomas T, Biesecker LG, Smith P,Fryer A, Chandler KE, Kerr B,Tassabehji M, Lynch SA,Krajewska-Walasek M, McKee S, Smith J, Sweeney E,MansourS, Mohammed S, Donnai D, Black G.Whole-exome-sequencing identifies mutations inhistoneacetyltransferase gene KAT6B in individuals with theSay-Barber-Bieseckervariant of Ohdo syndrome. Am JHum Genet, 2011, 89(5): 675–681.
[2] Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, Guo SL, He J, Chen YF, ZhangQJ, Li HF, Lin Y, Murong SX, Xu JF, Wang N, Wu ZY. Exome sequencing identifies truncatingmutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet,2011, 43(12):1252–1255.
[3] Hoischen A, van Bon BW, Rodríguez-Santiago B, Gilissen C, Vissers LE, deVries P, Janssen I, van Lier B, Hastings R, Smithson SF, Newbury-Ecob R,Kjaergaard S, Goodship J, McGowan R, Bartholdi D, Rauch A, Peippo M, Cobben JM,Wieczorek D, Gillessen-Kaesbach G, Veltman JA, Brunner HG, de Vries BB. De novononsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet, 2011,43(8): 729–731.
[4] Min BJ, Kim N, Chung T, Kim OH, Nishimura G, Chung CY, Song HR, Kim HW, LeeHR, Kim J, Kang TH, Seo ME, Yang SD, Kim DH, Lee SB, Kim JI, Seo JS, Choi JY, KangD, Kim D, Park WY, Cho TJ. Whole-exome sequencing identifies mutations of KIF22in spondyloepimetaphyseal dysplasia with joint laxity, leptodactylic type. Am JHum Genet, 2011, 89(6): 760–766.
[5] Zeitz C, Jacobson SG, Hamel CP, Bujakowska K, Neuille M, Orhan E, ZanlonghiX, Lancelot ME, Michiels C, Schwartz SB, Bocquet B, Congenital Stationary Night
Blindness Consortium, Antonio A, Audier C, Letexier M, Saraiva JP, Luu TD,Sennlaub F, Nguyen H, Poch O, Dollfus H, Lecompte O, Kohl S, Sahel JA,Bhattacharya SS, Audo I. Whole-exome sequencing identifies LRIT3 mutations as acause of autosomal- recessive complete congenital stationary night blindness.Am J Hum Genet, 2013, 92(1): 67–75
更多伯豪生物服務

